Unlocking Protein Function: New AI Method Predicts Biological Roles From Genetic Sequences
Invented by ADAMI; Christoph, C G; Nitash, HINTZE; Arend, Board of Trustees of Michigan State University
In today’s fast-moving world of science and technology, predicting what a piece of data means is very important. This is especially true when it comes to things like DNA strings, protein chains, and even patterns of brain activity. The patent application we will explore here introduces a new way to do just that—predict the function of any symbolic sequence using a special method called Information Decomposition for Sequences (IDSeq). Let’s break down what makes it special and how it could change the way we look at complex data.
Background and Market Context
Researchers and engineers often deal with long strings of symbols. In biology, these symbols could be the letters that make up DNA or proteins. In neuroscience, the symbols might show when a neuron fires in the brain. In medicine, the data could represent genetic codes linked to diseases. In all these fields, one big problem is figuring out what these sequences do—what is their function?
Predicting the function of a sequence is not just an academic challenge. It has huge value in real life. For example, if a doctor can quickly figure out if a gene causes a disease, treatment can start sooner. In drug discovery, knowing if a protein sequence leads to drug resistance can shape the next steps for new treatments. In AI, understanding how input patterns relate to output actions can make machines smarter and more reliable.
Current methods to uncover the meaning of these sequences often rely on building models or training algorithms. These approaches need large amounts of data so the model can “learn” the right patterns. However, real-world data is rarely perfect. There is always noise—random stuff that does not help but can confuse the model. With small or noisy datasets, these methods can make mistakes.
The demand for better, faster, and more accurate sequence analysis tools is growing. As more data becomes available (think of all the DNA sequencers running every day), there is a big market for smarter ways to analyze it. Health care, agriculture, drug development, and even artificial intelligence all benefit if we can crack the code of what these sequences mean.
This patent arrives in a world eager for solutions that do more with less: less data, less noise, and less need for complex training. If a method can predict the function of a sequence without heavy modeling, it opens the door for new discoveries, better diagnostics, and smarter AI.
Scientific Rationale and Prior Art
To understand what this patent brings, it helps to know what has been tried before. Traditional approaches to predicting the function of a sequence look for patterns. For example, in DNA research, scientists align many similar sequences and look for spots where certain letters (A, T, C, G) are common. They compare these spots across healthy and sick individuals to find links.
Many existing methods use a model-based approach. This means they try to build a mathematical or statistical model that describes the relationship between the sequence (input) and its function (output). These models are often trained using lots of known data. The catch? If the training data is small or noisy, the model can get things wrong. It might learn patterns that are just random, not real, and this leads to bad predictions.
One popular tool in biology is the Position Weight Matrix (PWM). A PWM counts how often each symbol (like a letter in DNA) appears at each position in a sequence. By comparing these counts, researchers can spot places that are important for function. However, most uses of PWMs look at one symbol at a time (first-order), not at pairs or triples of symbols (higher-order). This means they might miss more complex patterns.
Another idea from information theory is that the information in a sequence can be broken down, or decomposed, into contributions from single symbols, pairs, triples, and so on. This is called the information decomposition theorem. The more of these pieces you look at, the better you can describe the sequence—if you have enough data. But if you try to use too many pieces with too little data, you can end up just modeling noise.
Prior art often struggles with these trade-offs:
- Most methods either look at simple patterns (single symbols) or require big data to see more complex ones.
- Handling noise is tough—models often capture noise as if it were real signal.
- Many approaches do not generalize well outside biology (e.g., to neural data or other alphabets).
What has been missing is a simple, flexible method that can adapt to different types and sizes of data, works across domains, and can pick the right level of complexity based on the data at hand.
Invention Description and Key Innovations
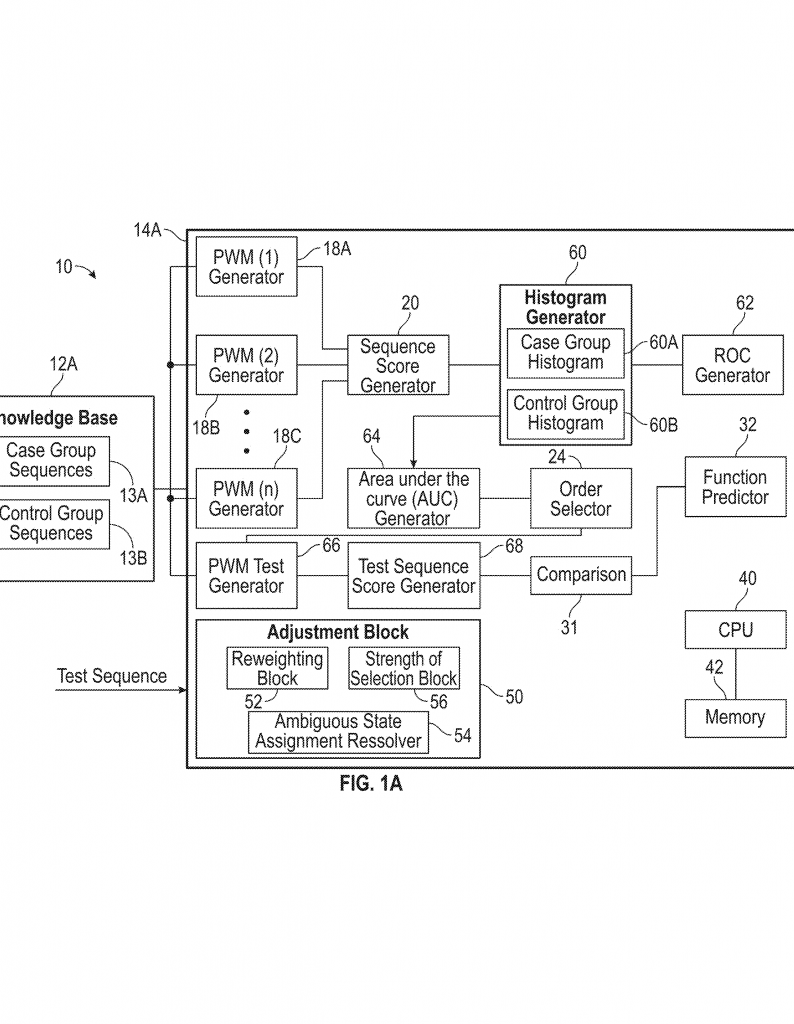
This patent introduces a new way to predict the function of a sequence, called Information Decomposition for Sequences (IDSeq). The main idea is to use information theory to break down the sequence into meaningful parts and calculate scores that relate to its function, without building a full model or heavy training.
Let’s walk through how it works in plain words:
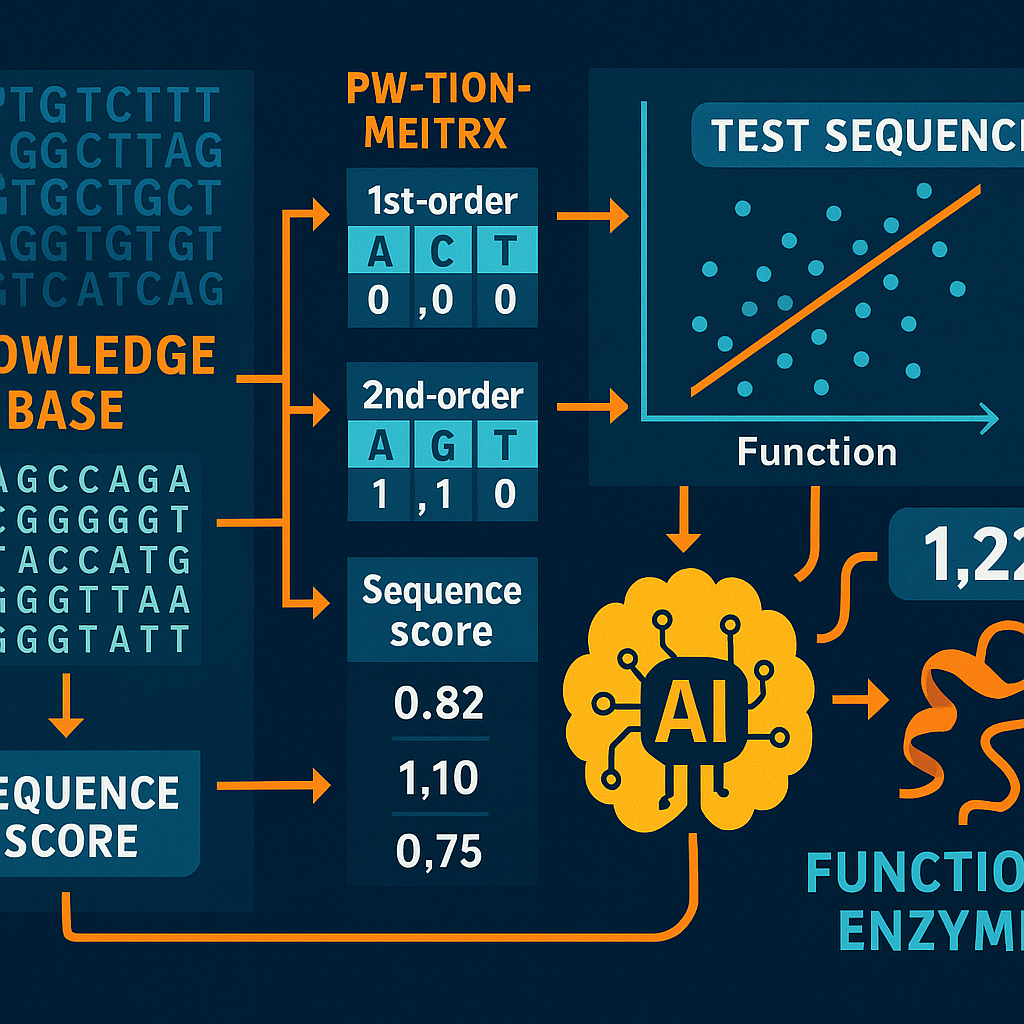
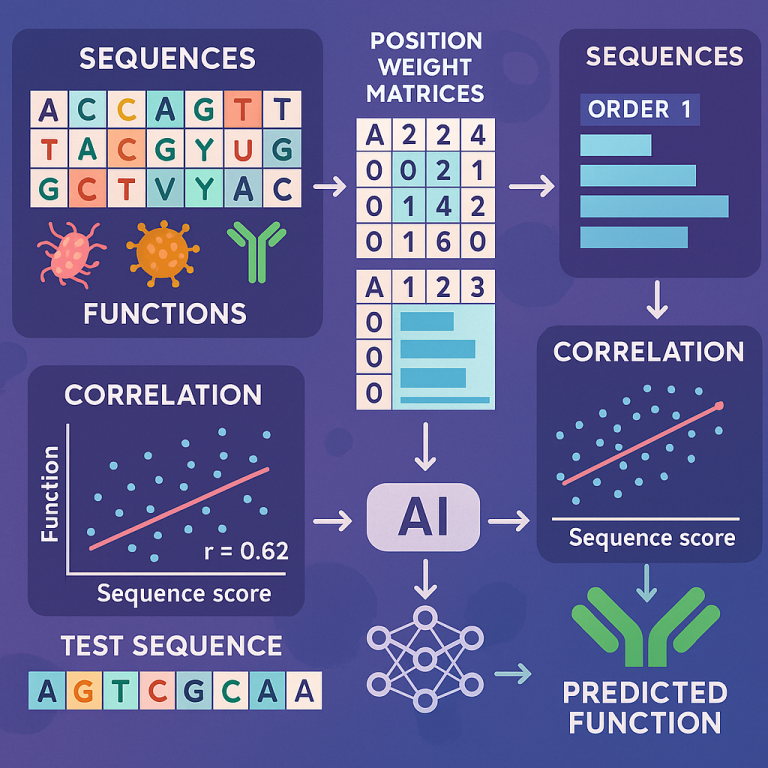
First, the system needs a knowledge base. This is a collection of many sequences where the function is known. For example, you might have a list of DNA sequences and know which ones are linked to a disease or how well they work in a lab test. These sequences could be from proteins, DNA, brain signals, or really any kind of symbol sequence.
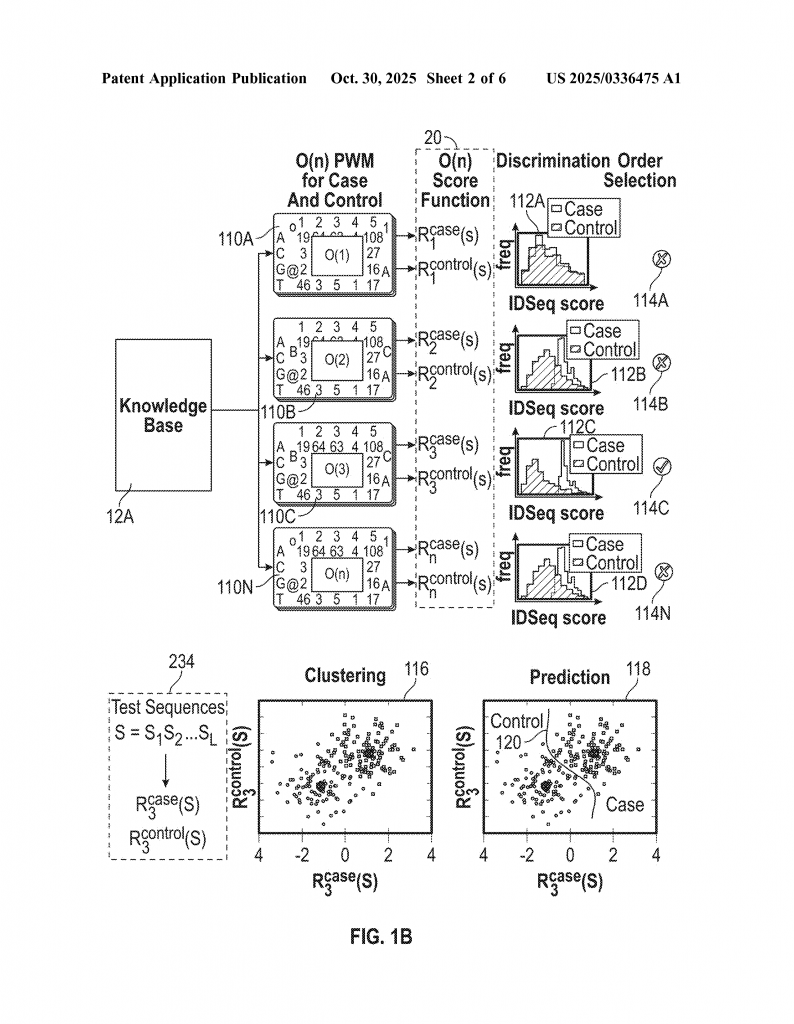
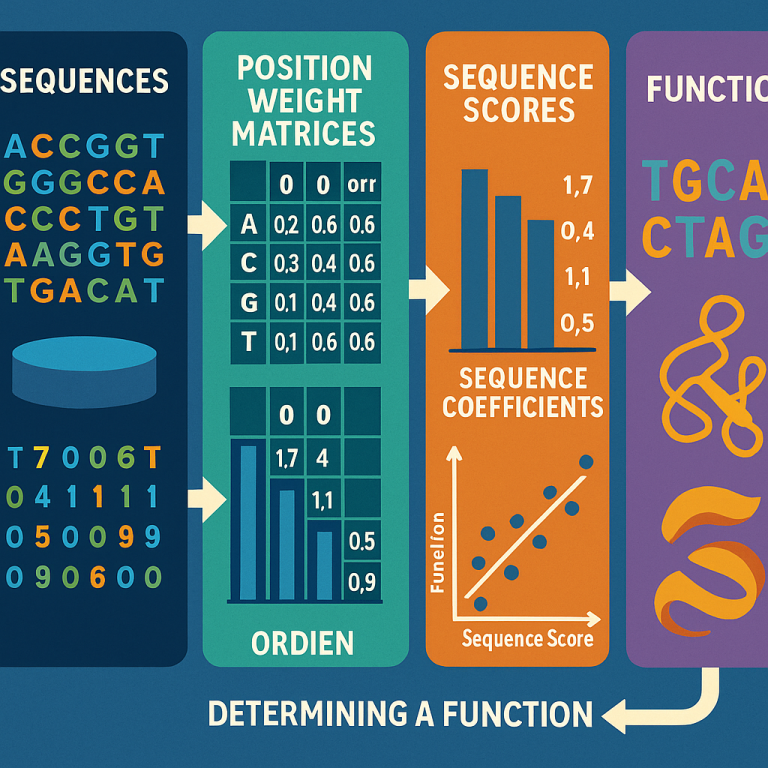
Next, the system builds Position Weight Matrices (PWMs) of different orders. A first-order PWM looks at single symbols at each spot in the sequence. A second-order PWM looks at pairs of symbols at two spots. A third-order PWM looks at triples, and so on. For example, in DNA, a first-order PWM would see how often an “A” shows up at the first position, a “T” at the second, and so on. A second-order PWM would look at how often an “A” at the first position is followed by a “G” at the second, and so forth.
The system then uses these PWMs to score each sequence in the knowledge base. The score tells how much information from the sequence matches the patterns found in the knowledge base. The more the sequence fits the patterns, the higher the score.
Now, for each sequence, the system checks how well these scores line up with the known function. It calculates something called a correlation coefficient—a number that shows how closely the score and the function are related. It does this for each order of PWM (first-order, second-order, third-order, etc.).
The key is to pick the “order” (first, second, third, etc.) where the score best matches the function. If the data set is small, maybe only first-order patterns are reliable. If there is more data, the system can safely use higher-order patterns to get more detail.
Once the best order is chosen, the system can score any new test sequence. By comparing the score of the new sequence to those in the knowledge base, it predicts the function of the new sequence. If the function is a number (like how well a protein works), the system can use regression—a way to map the score to the function value. If the function is a class (like disease or no disease), the system can use the scores to classify the new sequence.
The invention also includes ways to make the method more fair and accurate. For example, if some sequences in the knowledge base are very similar because they come from the same ancestor, the system can down-weight these to avoid bias. If there is uncertainty about what symbol is at a given spot (maybe because of errors in measurement), the system can split the weight between possible options. There are also ways to adjust how strongly the function influences the weight, so that the method matches the real-world process being studied.
A unique part of this invention is that it does not need to build a full mathematical model of the data. It simply counts patterns, calculates scores, and relates those scores to function. This makes it much faster and less likely to be fooled by noise. It can work with any kind of symbolic sequence, not just DNA or proteins, so it is very flexible.
The system can be used in many ways. It can classify sequences into groups (like healthy vs. sick), predict how well a sequence will perform a certain function, and more. The method can be run by a computer, and it can be built into software, hardware, or both.
In summary, the key innovations are:
- Building PWMs of any order (single symbols, pairs, triples, etc.) and picking the best order based on data.
- Using information decomposition to extract only meaningful patterns, ignoring random noise.
- Adapting the method to any kind of symbolic sequence and any function (number or class).
- Providing ways to adjust for common ancestry, ambiguous data, and strength of selection.
- Working without heavy modeling or training—just using what is in the data.
This makes the IDSeq system a powerful new tool for anyone who needs to figure out what a sequence means, in almost any field.
Conclusion
The patent for Information Decomposition for Sequences (IDSeq) offers a simple, flexible, and powerful method for predicting the function of any symbolic sequence. By breaking down information into manageable pieces and focusing on patterns with real support in the data, this approach avoids many of the pitfalls of current model-based methods. Its ability to adapt to different problems, ignore noise, and work without large amounts of training data makes it a valuable tool across biology, medicine, neuroscience, and even artificial intelligence. As more data becomes available and the need for fast, reliable function prediction grows, this invention stands ready to meet the challenge and drive new discoveries.
Click here https://ppubs.uspto.gov/pubwebapp/ and search 20250336475.

