Invention for Inhibitors for histone deacetylases and lysine specific desmethylases (HDACS),
Invented by Philip Cole, Shonoi Ming, Polina Prusevich, Jay Kalin, Johns Hopkins University
The dysregulation of HDACs and KDMs has been implicated in various diseases, including cancer, neurodegenerative disorders, and inflammatory conditions. As a result, there has been a growing interest in developing inhibitors that can modulate the activity of these enzymes. Inhibitors for HDACs and KDMs have shown promising therapeutic potential, making them an attractive target for drug development.
The market for inhibitors of HDACs and KDMs has witnessed significant growth in recent years. This can be attributed to several factors, including the increasing understanding of the role of these enzymes in disease progression, advancements in drug discovery technologies, and the need for more effective and targeted therapies.
In the field of cancer research, HDAC inhibitors have gained considerable attention. These inhibitors have been shown to induce cell cycle arrest, promote apoptosis (programmed cell death), and inhibit angiogenesis (the formation of new blood vessels). Several HDAC inhibitors have been approved by regulatory authorities for the treatment of hematological malignancies, such as cutaneous T-cell lymphoma and multiple myeloma. Additionally, clinical trials are underway to evaluate the efficacy of HDAC inhibitors in various solid tumors, including breast, lung, and prostate cancers.
Similarly, KDM inhibitors have shown promise in the treatment of cancer and other diseases. By targeting specific KDMs, these inhibitors can modulate gene expression patterns, leading to altered cellular functions. For instance, inhibitors of KDM4, a family of lysine-specific demethylases, have demonstrated anti-cancer effects by suppressing the growth and metastasis of tumor cells. Furthermore, KDM inhibitors have shown potential in the treatment of neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases, by modulating the expression of genes involved in neuronal function and survival.
The market for HDAC and KDM inhibitors is expected to continue growing in the coming years. The increasing prevalence of cancer and other diseases, coupled with the need for more targeted therapies, provides a favorable environment for the development and commercialization of these inhibitors. Moreover, advancements in drug delivery systems and personalized medicine approaches are expected to further enhance the efficacy and safety of HDAC and KDM inhibitors.
However, challenges remain in the development of HDAC and KDM inhibitors. One major hurdle is the identification of selective inhibitors that can specifically target individual HDAC and KDM isoforms, as these enzymes have diverse functions and substrates. Additionally, the potential side effects and toxicity profiles of these inhibitors need to be carefully evaluated to ensure their safety in patients.
In conclusion, the market for inhibitors of HDACs and KDMs is witnessing significant growth due to their potential therapeutic applications in various diseases. The development of selective and safe inhibitors holds great promise for improving patient outcomes and addressing unmet medical needs. Continued research and investment in this field are essential to unlock the full potential of HDAC and KDM inhibitors in the treatment of diseases.

The Johns Hopkins University invention works as follows
The present invention is a series of phenelzine analogues that contain a phenelzine molecule linked to a aromatic moiety, and are used as inhibitors of the lysine specific demethylase 1 enzyme (LSD1) or one or more histone acetylases. The phenelzine analogues disclosed herein exhibit potency and selectiveness for LSD1 enzymes versus MAO or LSD2 and also exhibit gene-specific histone methylation, anti-proliferation activity in cancer cell lines and neuroprotection when exposed to oxidative stresses. The phenelzine analogues disclosed herein can be used for treating diseases, conditions or disorders that are related to LSD1 or HDACs. This includes, but is not limited to cancers and neurodegenerative disease.

Background for Inhibitors for histone deacetylases and lysine specific desmethylases (HDACS),
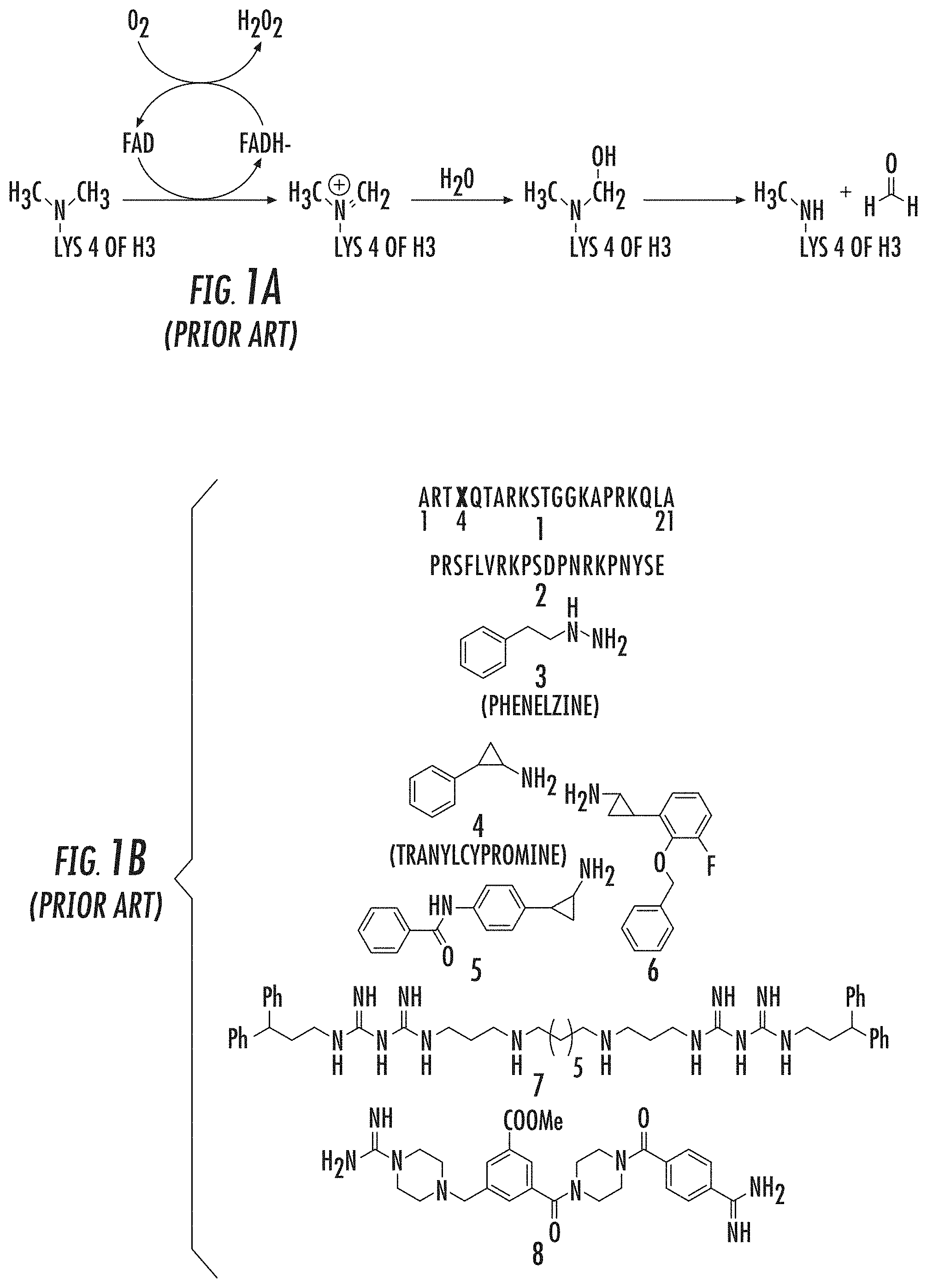
Reversible histone-lysine methylation” is a key mechanism in regulating gene expression and chromatin dynamics. Lysine specific demethylase 1, the first histone-demethylase discovered, is responsible for oxidatively breaking one or two methyl group from Lys4 on histone H3(H3K4). Culhane J. C. and Cole P. A. (2007). LSD1 may play a part in gene silencing in this way. This is because methylation H3K4 at promoters is an established chromatin marker linked to transcriptional activity. Liang, G., et al. (2004), Heintzman, N. D., et al. (2007).
Since its discovery, LSD1’s histone demethylase has been studied as a potential pharmacologic treatment for cancer and other illnesses. LSD1 levels have been found to be elevated in many cancers. Lv, T., et al. (2012), Lim, S., et al. (2010), Metzger, E., et al. (2005). Murray-Stewart T. et. al. suggest that LSD1 blockers could theoretically reactivate a number of tumor suppressors, which have been shown in cancer to be silenced by epigenetic processes. (2013), Huang, Y., et al. (2007), Huang, Y., et al. (2009), Jin, L., et al. 2013 as with DNA methyltransferase and histone deacetylase inhibitors. Takai N. and Narahara H. (2008).
LSD1 (a 90 kDa flavin bound enzyme) belongs to the superfamily of amine oxidase proteins. It uses molecular oxygen as a cosubstrate, and produces hydrogen peroxide as a byproduct. 1A). Culhane J. C. and Cole P. A. (2007), Shi, Y., et al. (2004), Gaweska, H. (2009), Forneris, F. (2005). LSD1’s enzyme mechanism does not allow it to demethylate H3K4Me3 trimethylated Lys4. However, members of iron-dependent Jmj demethylases can perform this function. Culhane J. C. and Cole P. A. (2007); Tsukada, Y., et al. (2005). “In addition to the C terminal amine-oxidase catalytic region, LSD1 contains an N-terminal SwIRM domain as well as a 105aa Tower domain which is located within the amine-oxidase-domain that can bind the CoREST.
Several LSD1 Demethylase Inhibitors have been reported. These include peptides (1-2), MAOIs, derivatives of MAOIs (3-6), Polyamines (7) and Guanidine-containing compounds ((8) (FIG. 1B). Yang, M., et al. (2007), Culhane, J. C., et al. (2010), Dancy, B. C. R., et al. (2012), Tortorici, M., et al. (2013), Binda, C., et al. (2010), Mimasu, S, et al. (2010), Zhu, Q., et al. (2012), Wang, J., et al. (2011), Culhane, J. C., (2006), Pollock, J. A., et al. (2012), Gooden, D. M., et al. (2008) Dulla, B., et al. (2013), Hazeldine, S., et al. (2012).
The development of tranylcypromine analogues has proven to be a promising strategy. Pollock, J. A., et al. (2012), Gooden, D. M., et al. (2008). Tranylcypromine, a MAO inhibitor, is used to treat clinical depression. It is weakly effective as an LSD1-based mechanism-based inactivator. (Ki(inact), 0.5 mM; k(inact), 0.67 min?1). Yang, M., et al. Schmidt, D. M. Z. and McCafferty D. G. (2006). However, it has been demonstrated that the addition of a aryl to tranylcypromine could produce more potent LSD1 inhibitors. Binda, C., et al. (2010), Mimasu, S, et al. (2010). Further, phenelzine (a MAO-inhibitor used to treat depression) was shown to be stronger than tranylcypromine in inhibiting LSD1. Culhane, J. C., (2010)
In certain aspects, the subject matter disclosed herein provides a compound of Formula I:
wherein:
t” is an integer chosen from the range of 0, 1, 2, 3 and 4;
L is the linking group that was selected from a group consisting?X1′,? [X1?C(?O)?NR1]d?, ? [X1?NR1?C(?O)]d?, ? [C(?O)?NR1?X1]d?, ? [NR1?C(?O)?X1]d?, ? [NR1?C(?O)?NR1?X1]d?, ? [X1?NR1?C(?O)?NR1]d?, ? [X1?O?C(?O)?NR1]d?, ? [O?C(?O)?NR1?X1]?, ? [X1?NR1?C(?O)?O]d?, ? [NR1?C(?O)?O?X1]d?, ?X1?O?, ?X1?NR1, ?X1?S?, ?X1?SO?, ?X1?SO2?, ?X1? O? X1?, ?X1? NR1? X1?, ?X1? S? X1?, ?X1?SO? X1? SO2? “X1? where d is a number selected from the range of 1, 2, 3 and 4;
where X1 is chosen from the group consisting? (CH2)n?, ? [(CH2)n?CH?CH? (CH2)m]e?, ? [(CH2)n?C?C? ? The? (CH2)n?, ? The? (CH2)n?,? The?CH?CH. (CH2)n?and ? (CH2)m? (CH2)m? (CH2)n? (CH2)n? (CH2)m? (CH2)m?

R1 and R’1 are independently selected from the following group: hydrogen, substituted and unsubstituted linear and branched alkyls, alkoxyls, substituted and unsubstituted substituted and unsubstituted homoaryls, substituted and unsubstituted substituted and unsubstituted substituted and unsubstituted substituted and unsubstituted substituted aryls, substituted and unsubstit
(CH2)p?NR3?NR4R5 or?? “R2 is? The? (CH2)p? (CH2)p? (CH2)p? (CH2)p?
Each R?2 at each occurrence is selected independently from the group consisting allyls, hydroxyls, carboxyls, aminos, halogens, nitros, oxos,?CF3, substituted and unsubstituted anaryls, substituted and unsubstituted homoaryls;
R3, R4, R5, and R24 are independently selected from the following group: hydrogen, substituted, unsubstituted linear, branched, or cycloalkyls, substituted, unsubstituted homoaryls, substituted, unsubstituted substituted aryls, substituted, unsubstituted substituted heteroaryls, and R24 can be substituted, unsubstituted, linear, or branched, unsubsti
X2 is chosen from the group consisting halogens,?O?Si (R21R22),2?R23 wherein each R21,R22,and R23 are independently substituted or unsubstituted linear alkyls;
A” is chosen from the following group: substituted and unsubstituted monocyclic cycloalkyls, substituted and unsubstituted multicyclic cycloheteroalkyls, substituted and unsubstituted homoaryls, substituted/unsubstituted substituted aryls, substituted/unsubstituted substituted arylalkyls, or substituted/unsubstituted polycyclic aryl
where one or more carbons atoms in ring B may be replaced by one or two heteroatoms from the group of N, S, and O;
wherein either one or both ring structures B and A can be substituted optionally with one or more groups reactive capable of forming prodrugs;
and pharmaceutically accepted salts, hydrates and solvates of the same.
The compound Formula (I), in particular, has the following structure.

Click here to view the patent on Google Patents.

